C3.3: Computation of Electronic and Intermolecular Interactions
Subproject Leader: Wim Klopper
Institut für Physikalische Chemie, KIT
Contributing Scientists:
Present: Angela Bihlmeier, Nils Middendorf, Konstantinos Vogiatzis
Past: Robert Barthel, Stephan Bernadotte, Florian Bischoff, Karin Fink
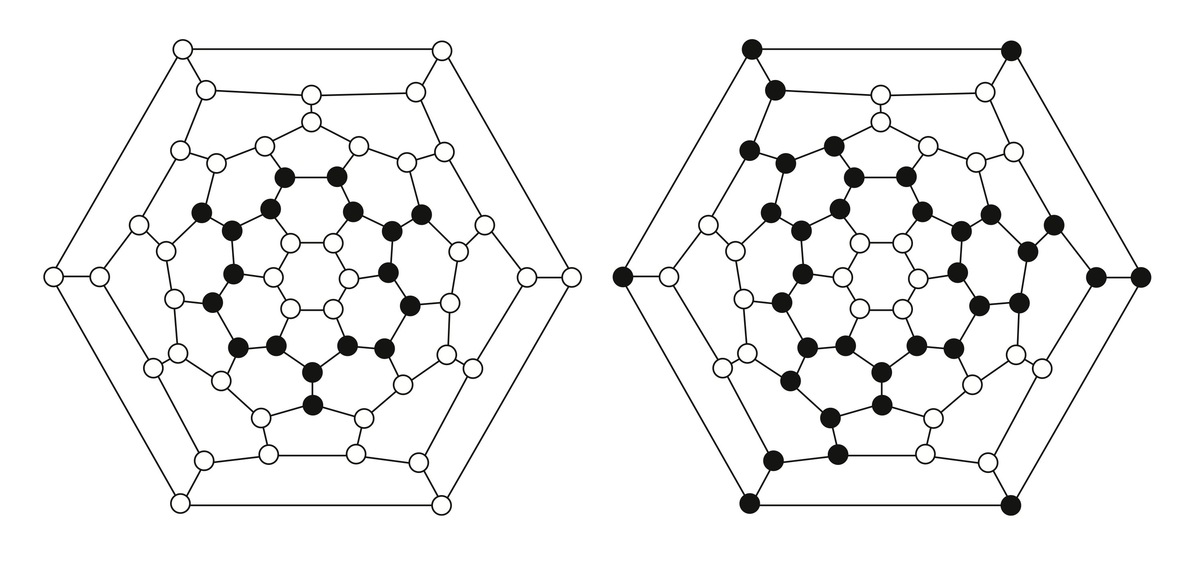
1. Schlegel diagrams of C60H18 and C60H36

Subtle Effects Require Predictive-Level Theory
Subproject C3.3 focuses on the computation of electronic and intermolecular interactions in molecule-based nanostructures. Such computations are crucial for the understanding of the (perturbed) properties of the molecules (e.g., structural parameters, binding energies, optical, magnetic, and electrical properties) and for the design of new functional nanostructures. Often, the effects are subtle, and for molecular systems on the nanoscale, the accurate and thus reliable ab initio calculation of such effects is a challenging task requiring continuous method development.
Carbon-Based Nanostructures
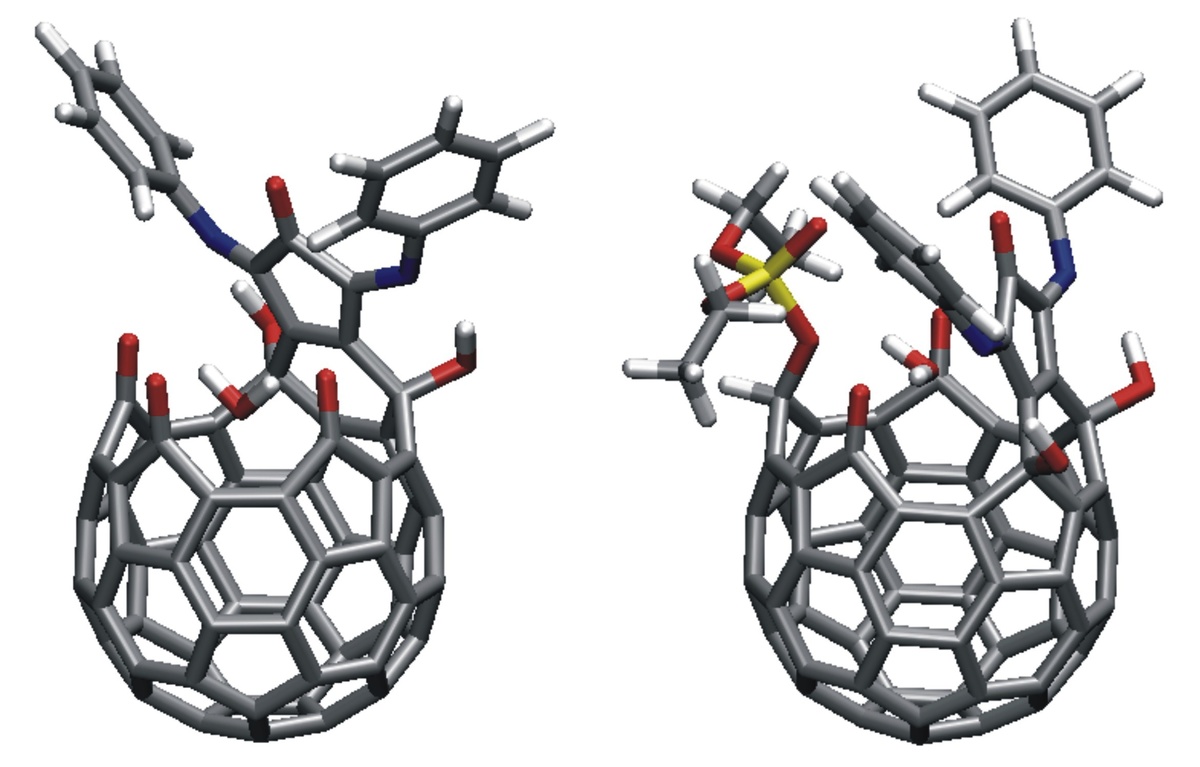
Motivated by experiments in subproject C4.6 on the exposure of thin fullerene films to atomic deuterium, fullerene–fullerene interactions are modeled. Inter-cage binding occurs for fullerenes that do not obey the isolated-pentagon rule (IPR), such as C58 or C60(C2v). In contrast, the common IPR-obeying fullerene C60(Ih) shows only weak van der Waals inter-cage interactions. DFT calculations on the hydrogenation of various fullerenes (e.g., C50, C58, C60) predict the energies and molecular structures of a variety of hydrogenation products such as C60H18 and C60H36 (Figure 1) [1]. In other work on fullerene derivatives, structures and desorption energies of anions of switchable water-encapsulating open-cage fullerenes are studied. Transition-state structures and barrier heights for water loss/gain are obtained (Figure 2) [2]. The results are similar to the corresponding data for the neutral cages as obtained from DFT calculations and from NMR measurements of H2O/D2O-exchange rates. An easily removable phosphate moiety serves as the “cork” to close the “molecular bottle”.
Magnetic Exchange-Coupling Constants
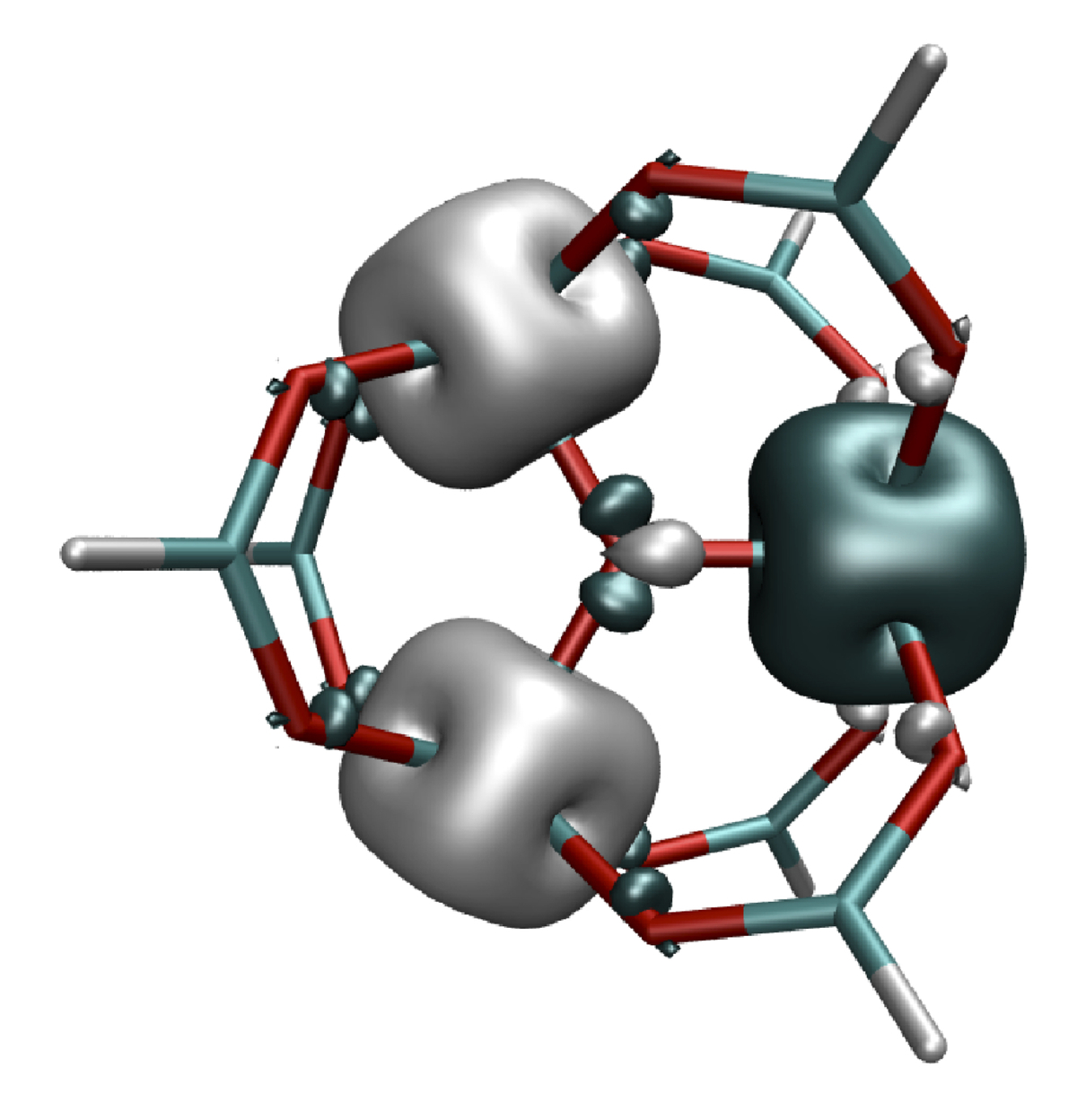
At the DFT level, the Noodleman approach with symmetry-broken determinants (Figure 3) is applied to compute magnetic exchange-coupling constants of single-molecule magnets (see subproject C1.2). DFT results are compared with those obtained at the multi-reference configuration-interaction level. Magnetic susceptibilities are computed and compared to experimental data. The calculations confirm the known strong dependence of broken-symmetry-DFT calculations on the functional used while a good agreement with experiment is often achieved by a modified configuration interaction (MCI) approach that has been designed specifically for the purpose of computing magnetic exchange-coupling constants [3].
Unconventional Bonding Situations
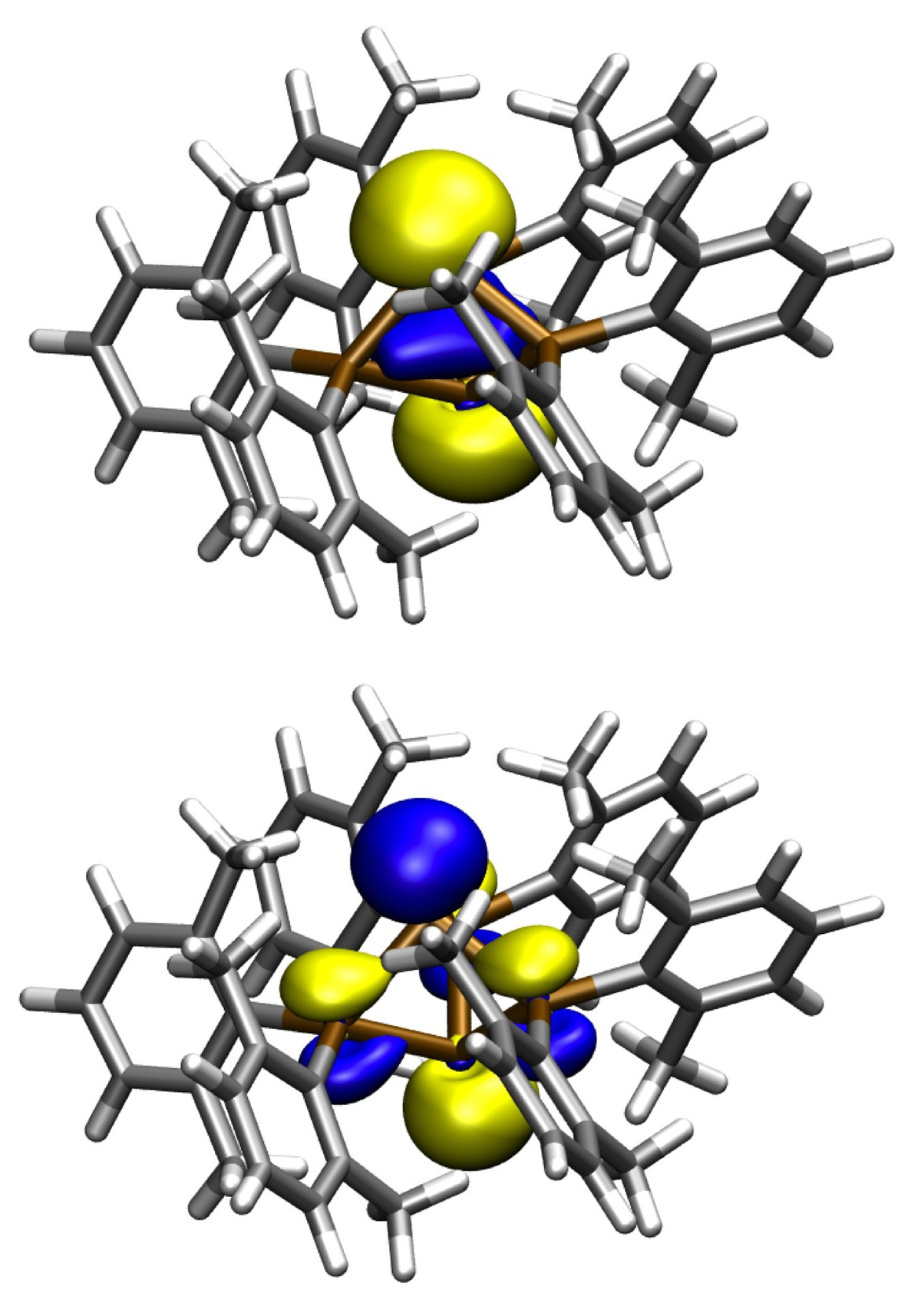
Unusual bonding occurs between bridgehead atoms in propellanes (Figure 4) and related larger clusters. The formation of a singlet hexaradicaloid metalloid Ge14 cluster from three singlet biradicaloid fragments has been studied in collaboration with subproject C1.3 [4]. The metalloid clusters may be viewed as model compounds for the area between molecules and the solid state. Hence, the observed multi-radicaloid character in the Ge14 cluster may be of importance for nanoparticles and surfaces where unsaturated Ge atoms are found. The multi-radicaloid character may be the reason for the differences in physical and chemical properties between nanoparticles and the bulk phase, for example, the reason for the a strong size-dependent photoluminescence (PL) of Ge nanoparticles versus no comparable PL in elemental germanium.
Intermolecular Complexes
Motivated by experimental work in subproject C5.2 (work directed towards the synthesis of functionalized organic nanostructures including systems containing six [2]pseudorotaxanes arranged octahedrally around a C60 fullerene), DFT calculations are performed on various [2]pseudorotaxanes built from a crown ether and the dibenzylammonium cation (Figure 5). The dibenzylammonium–crown ether [2]pseudorotaxanes are particularly interesting from a computational point of view, because they have much conformational freedom, and because they display competing intermolecular interactions such as N+-H…O hydrogen bonds versus van der Waals-type π-π stacking interactions.
References
|
[1] |
A. Bihlmeier and W. Klopper, Phys. Chem. Chem. Phys. 11, 1050 (2009) |
|
[2] |
O. Hampe, T. Karpuschkin, M. Vonderach, P. Weis, Y.M. Yu, L.B. Gan, W. Klopper, and M.M. Kappes, Phys. Chem. Chem. Phys. 13, 9818 (2011) |
|
[3] |
H. Fliegl, K. Fink, W. Klopper, C.E. Anson, A.K. Powell, and R. Clérac, Phys. Chem. Chem. Phys. 11, 3900 (2009) |
|
[4] |
C. Schenk, A. Kracke, K. Fink, A. Kubas, W. Klopper, M. Neumaier, H. Schnöckel, and A. Schnepf, J. Am. Chem. Soc. 133, 2518 (2011) |
|
[5] |
M. Klipfel, C. Réthoré, T. Muller, S. Bräse, and W. Klopper, Comput. Theor. Chem. 966, 186 (2011) |
List of Publications 2006-2011 as PDF
Subproject Report 2006-2010 as PDF