E4.1: Super-Resolution Optical Microscopy
Subproject Leader: Ulrich Nienhaus
Contributing Scientists:
Present: Stefan Brandholt, Rene Dörlich, Per Niklas Hedde, Annika Hense, Andrei Kobitski, Pauline Maffre, Karin Nienhaus, Florian Stockmar
Past: Jochen Fuchs, Susan Gayda (Böhme), Michael Herrman, Robert Rieger, Hendrik Sielaff

Imaging Beyond Abbe’s Diffraction Barrier
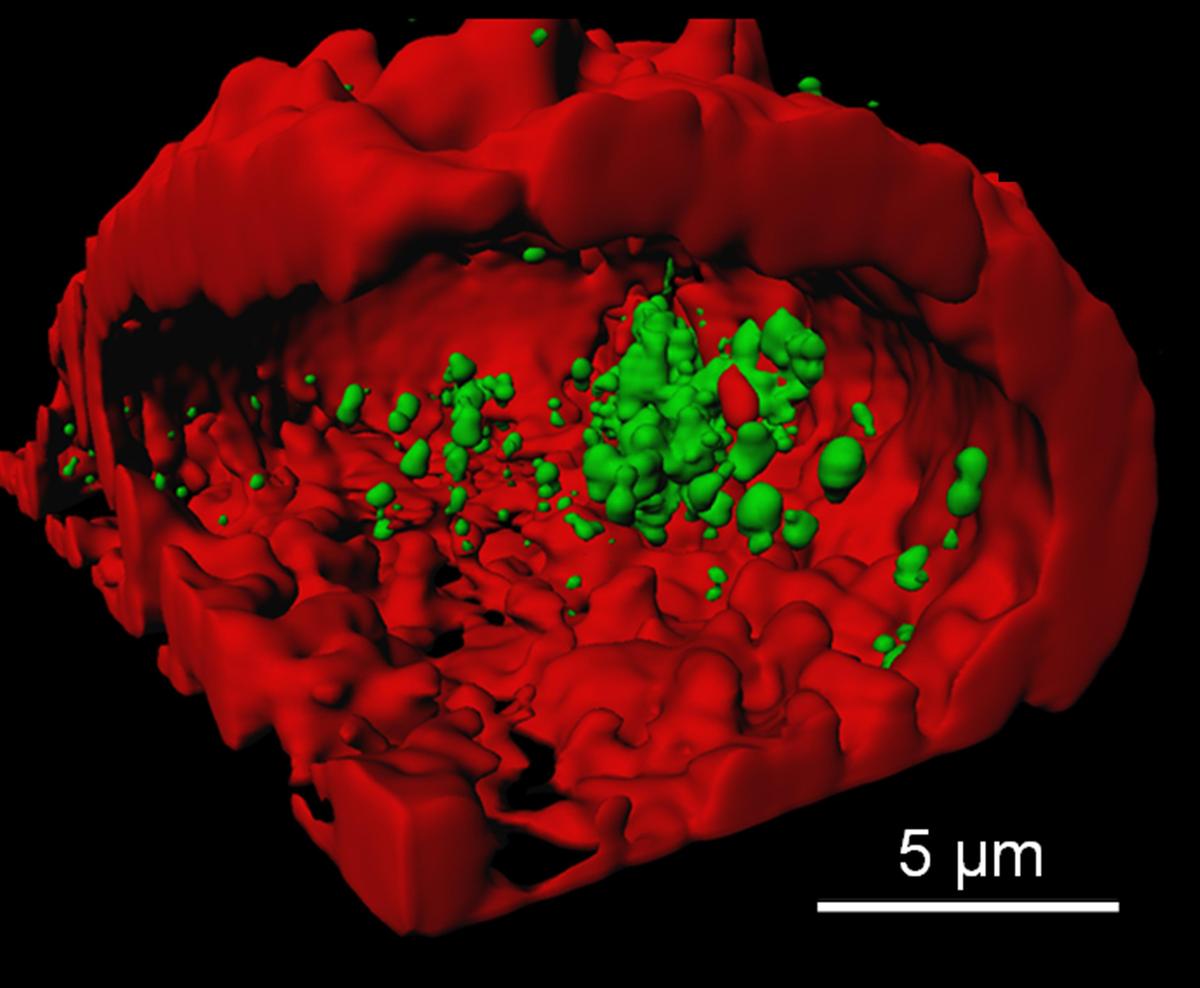
Light microscopy is arguably the most important technique for the study of living systems because it allows cells and tissues to be imaged in all three spatial dimensions over extended periods of time under minimally invasive, close-to physiological conditions. Conventional light-optical microscopy is diffraction-limited and, hence, only structures larger than ~200 nm can be resolved. Here we develop and apply super-resolution techniques for live-cell imaging. We have built a wide-field microscope for localization microscopy (PALM, STORM) featuring a resolution of ~20 nm when imaging photoactivatable fluorescent proteins. We also have set up a STED microscope for imaging and fluorescence correlation spectroscopy (FCS). For live-cell imaging of fast dynamics, further advances are needed to speed up data acquisition.
Online Image Analysis – What You See What You Get
Localization microscopy is based on the detection of individual fluorophores that can be sparsely photoactivated (‘switched on’) so that their signals do not overlap and their locations can be precisely determined. It is essentially an image processing technique, based on the reconstruction of high-resolution images from a few hundred up to a few thousand CCD camera frames of a wide-field microscope. We are pursuing two strategies for fast image reconstruction, namely (1) the use of ‘raw computation power’ by largely parallelized computation of the molecule loci in graphics processing units, and (2) the use of fast, algebraic localization algorithms that avoid the tedious fitting procedure [1]. Ongoing improvements of the image analysis include robust and efficient algorithms for the treatment of overlapping signals and the tracking of individual fluorophores over time.
Advanced Luminescent Markers for Super-Resolution Optical Imaging
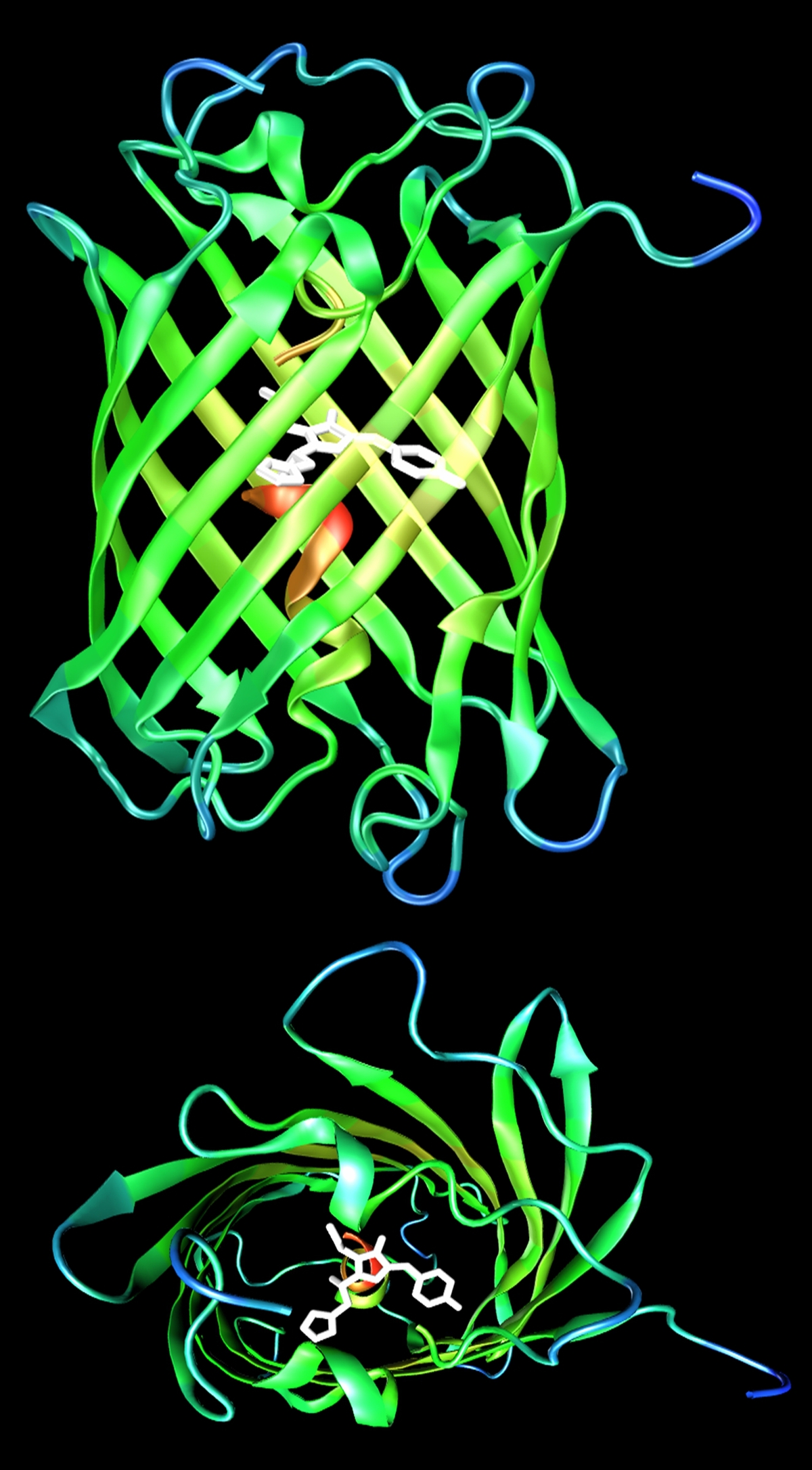
All super-resolution imaging relies on the capability to control the emission of fluorophores by light irradiation. While STED microscopy uses a fundamental physical mechanism, i.e., stimulated emission, the other super-resolution techniques utilize special photoactivatable fluorophores the emission of which can be controlled by light. Photoactivatable fluorescent proteins are popular markers for studies in living cells and organisms because they are genetically encoded and thus are produced by the systems under investigation themselves; no labeling steps are required. We have recently introduced IrisFP [2,3], a fluorescent protein offering two photoactivation modes: It is a so-called photoswitchable green fluorescent protein and can be switched between a fluorescent and a non-fluorescent state. After exposure to 400 nm light, IrisFP exhibits irreversible photoconversion to a photoswitchable red fluorescent protein. Its two photoactivation modes allow entirely new experimental approaches, e.g., pulse-chase imaging with sub-diffraction resolution in living cells by using dual-color PALM. Further improvements of these marker tools focus on color tuning and, most importantly, photostability issues. In addition, we also develop luminescent semiconductor and metal nanocrystals for application in live-cell imaging experiments [4].
Quantitative In-Vitro and In-Vivo Fluorescence Microscopy
Direct optical imaging of biomolecular dynamics in living cells is currently limited to a few milliseconds by CCD camera frame times. However, many interesting processes require a higher temporal resolution. To study faster dynamics, we perform particle tracking experiments or measure time correlations at fixed or slowly varying positions by FCS or two-focus-FCS (2fFCS). Alternatively, we analyze confocal images by raster image correlation spectroscopy (RICS).
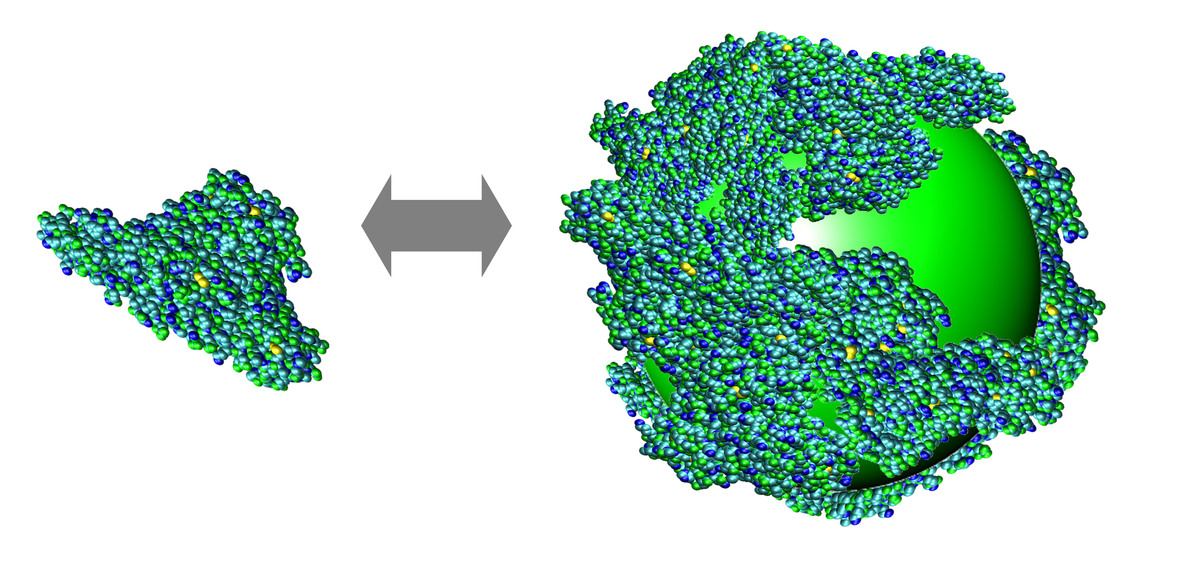
Using FCS in vitro, we were able to precisely monitor the adsorption of proteins onto small (10 – 20 nm diameter) nanoparticles [5]. The thickness of the forming protein monolayer is related to the molecular structures of the proteins, and the binding affinity can be understood in terms of electrostatic interactions. A quantitative understanding of the protein corona enshrouding the nanoparticle is of utmost importance for the safe application of nanoscale objects to living organisms.
Furthermore, we employ Förster resonance energy transfer (FRET) on FRET-labeled biopolymers in single-molecule in-vitro experiments to observe intramolecular conformational changes and bimolecular interactions. Efforts are currently ongoing to utilize FRET biosensors in the context of the living cell.
References
| [1] | P.N. Hedde, J. Fuchs, F. Oswald, J. Wiedenmann, and G.U. Nienhaus, Nature Methods 6, 689 (2009) |
| [2] | V. Adam, M. Lelimousin, S. Boehme, S., G. Desfonds, K. Nienhaus, M.J. Field, J. Wiedenmann, S. McSweeney, G.U. Nienhaus, and D. Bourgeois, Proc. Natl. Acad. Sci. USA 105 (2008) 18343-18348 |
| [3] | J. Fuchs, S. Böhme, F. Oswald, P.N. Hedde, M. Krause, J. Wiedenmann, and G. U. Nienhaus, Nature Methods 7, 627 (2010) |
| [4] | X. Jiang, C. Röcker, M. Hafner, and G.U. Nienhaus, ACS Nano 4 (2010) 6787-6797 |
| [5] | C. Röcker, M. Pötzl, F. Zhang, W. J. Parak, and Nienhaus, G. U. Nature Nanotechnology 4, 577 (2009) |
List of Publications 2006-2011 as PDF
Subproject Report 2006-2010 as PDF