C3.6: Theory of Transport through Single Molecules
Subproject Leader: Gerd Schön, Fabian Pauly (KIT Young Investigator Group Leader 2007-2011)
Contributing Scientists:
Present: Marius Bürkle, Thomas Hellmuth, Matthias Hettler (INT), Wolfgang Wenzel (INT)
Past: Michael Häfner, Janne Viljas, Juan Carlos Cuevas (Helmholtz Young Investigator Group Leader until 12.07)

Charge Transport at the Molecular Scale
In this project we aim at a better understanding of charge transport at the molecular scale. Chemical synthesis offers unique ways to control the molecular properties. It is our goal to develop theoretical tools for determining which realizations from the plethora of possibilities are most suited to achieve the desired functionalities or to describe novel phenomena arising at the nanoscale. Considering the atomic-scale junctions as building blocks of larger electric circuits, research results may lead to the further miniaturization of integrated circuitry, their improved performance, or new functionalities.
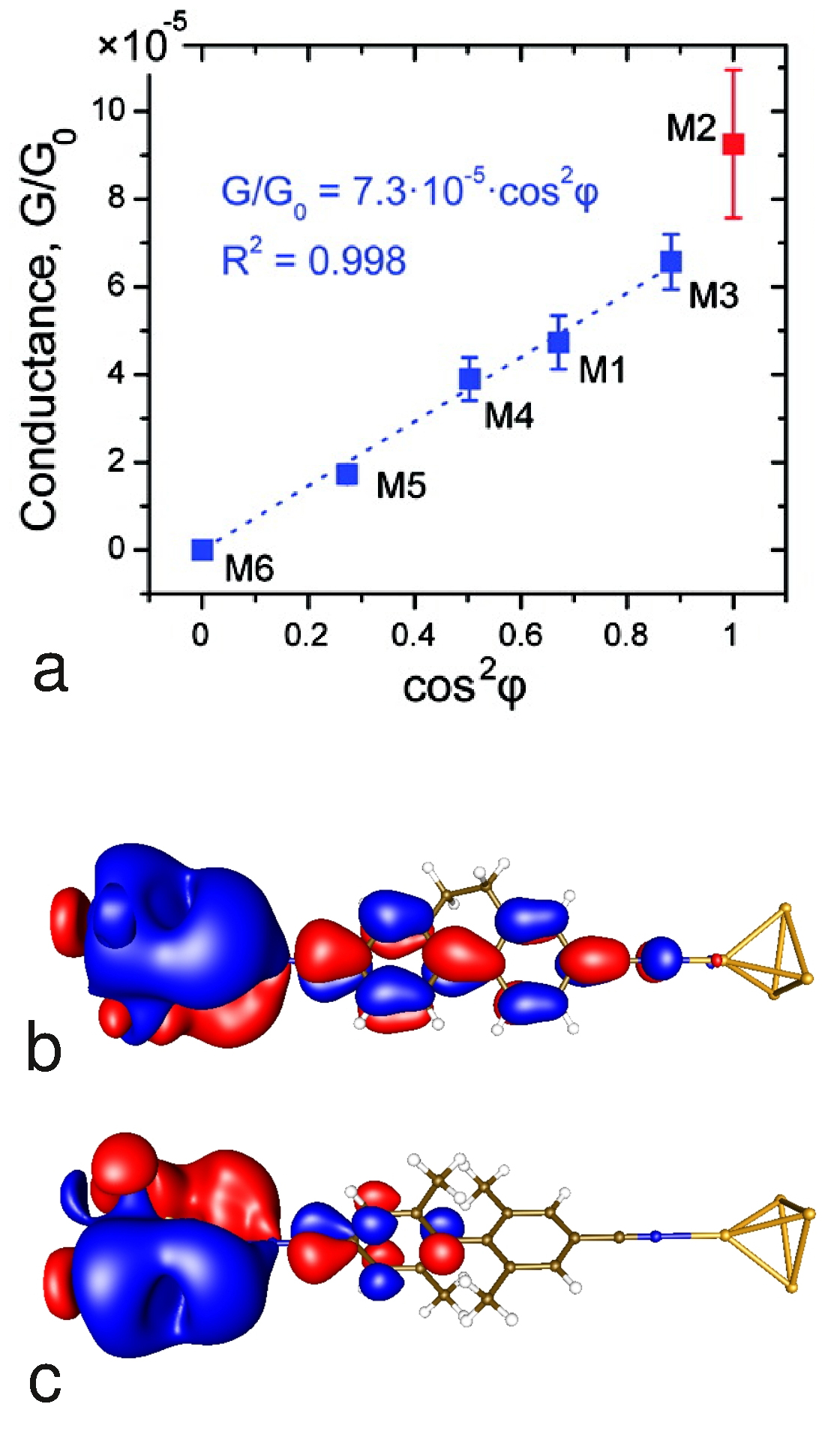
Conductance-Structure Relationships
Together with colleagues from the CFN we have analyzed how changes in the molecular structure are related to those in single-molecule junction transport. In particular, at the example of biphenyl molecules, we have studied the influence of conjugation on conductance [1]. Theory and experiment consistently show a cos2 φ dependence of the conductance on the torsion angle φ between the two phenyl rings. This confirms simple expectations based on the overlap of the π electron systems of the rings.
Alignment of Metallic and Molecular Levels
While the molecular structure is one important aspect determining the single-molecule conductance, the molecule-electrode interface is another. By changing the anchoring group, which establishes the contact to the metal, charge transport can for instance change from hole-dominated to electron-dominated [1]. While measurements of the electric current alone cannot distinguish between these two scenarios [2], this can be achieved by use of the thermopower [3].
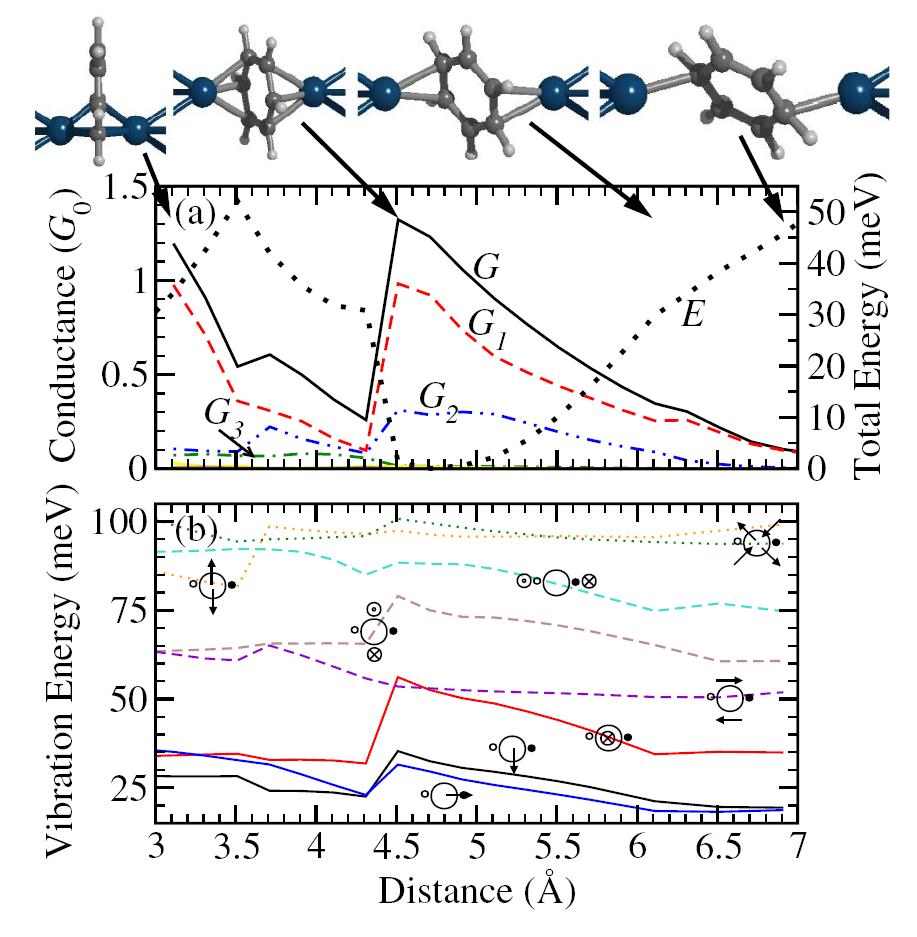
Under certain conditions it is also possible to drop the anchoring groups completely. For a benzene molecule contacted to platinum electrodes, we found that the direct binding via the molecular π system leads to highly conductive molecular junctions [4].
Further Collaborations within the CFN
Our atomistic ab-initio description of quantum transport [5] is based on the efficient implementation of density functional theory in TURBMOLE, as developed and maintained by our CFN colleagues from theoretical chemistry. In collaboration with them we have recently extended our elastic transport scheme to include also inelastic effects due to the excitation of vibrations. Beside the ab-initio scheme we also work with more empirical approaches to describe new physical effects or larger systems.
References
| [1] | A. Mishchenko, L. A. Zotti, D. Vonlanthen, M. Bürkle, F. Pauly, J. C. Cuevas, M. Mayor, and T. Wandlowski, J. Am. Chem. Soc. 133, 184 (2011) |
| [2] | L. A. Zotti, T. Kirchner, J. C. Cuevas, F. Pauly, T. Huhn, E. Scheer, and A. Erbe, Small 6, 1529 (2010) |
| [3] | F. Pauly, J. K. Viljas, and J. C. Cuevas, Phys. Rev. B 78, 035315 (2008) |
| [4] | M. Kiguchi, O. Tal, S. Wohlthat, F. Pauly, M. Krieger, D. Djukic, J. C. Cuevas, and J. M. van Ruitenbeek, Phys. Rev. Lett. 101, 046801 (2008) |
| [5] | F. Pauly, J. K. Viljas, U. Huniar, M. Häfner, S. Wohlthat, M. Bürkle, J. C. Cuevas, and G. Schön, New J. Phys. 10, 125019 (2008) |
List of Publications 2006-2011 as PDF
Subproject Report 2006-2010 as PDF