CFN – Theoretical Chemistry: Pathbreaking Review Paper
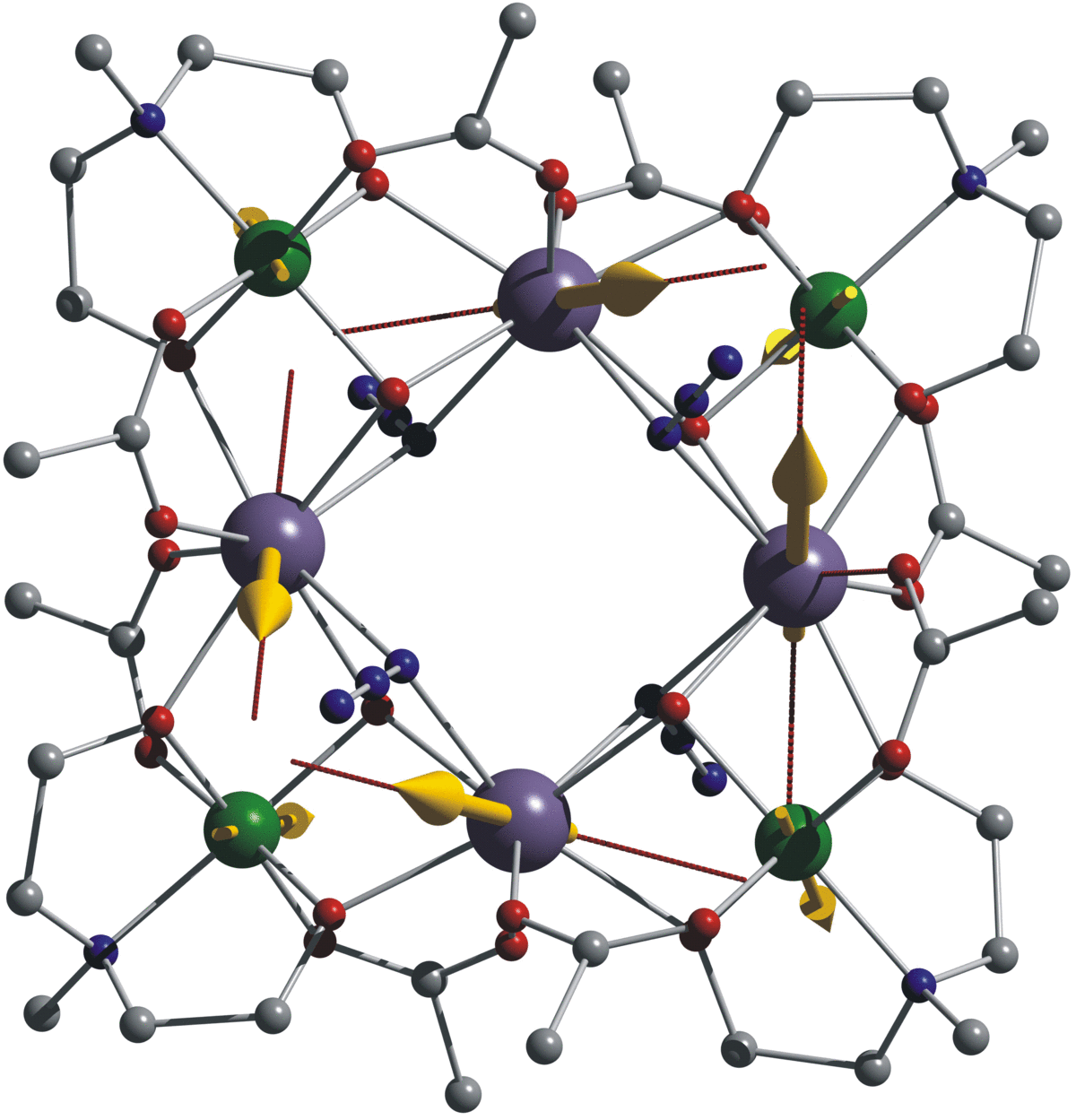
Density Functional Theory (DFT) for Open-shell Molecules: Spin Calculations
The review paper on the consideration of spin in density functional theory (DFT), published by the scientists Dr. Christoph Jacob from the Center for Functional Nanostructures (CFN) of Karlsruhe Institute of Technology and Professor Markus Reiher from ETH Zurich, functions to close a major knowledge gap in theoretical chemistry.
DFT is an important tool within the field of theoretical chemistry. It is used to calculate the properties of molecules and solids, such as binding lengths and energies.
For the time being, molecules with paired electrons only can be calculated using DFT. For molecules with unpaired electrons, also called open-shell systems, DFT cannot yet be applied in a satisfactory manner. Unpaired electrons lead to a magnetic moment, the spin, and its consideration by DFT remains quite challenging.
In transition metals and their compounds, these unpaired electrons offer a rich and complex chemistry, making them interesting for a number of applications. Change of spin in bio-inorganic reactions, for instance, may be responsible for a transition metal complex acting as a catalyst. Transition metal clusters in single molecule magnets are integral as connections of storage elements in (quantum) information processing.
The large scope of applications explains the high scientific interest in calculating the behavior of open-shell systems using theoretical methods. In past years, scientists Jacob and Reiher began systematic studies in this field and developed solution approaches. Currently, they have published their detailed results in the International Journal of Quantum Chemistry in the form of a tutorial that is highly useful for both beginners and advanced practitioners.
“Understanding of the underlying exact theory is a prerequisite for the development of a reliable approximation method. That is why we first sum up these fundamentals,” Jacob says. The authors explain the non-relativistic treatment of spin for a single electron – the subject of many basic studies in quantum mechanics. They then analyze the treatment of spin using the Hohenberg-Kohn and Kohn-Sham DFT. Various methods of considering spin in DFT are described and analyzed. Jacob and Reiher also study the relativistic DFT. Finally, proposals are made as to how spin approximation can be improved within the DFT.
Density Functional Theory (DFT)
In the 1960s, fostered by the newly emerging computer generation, quantum chemistry developed into its own field of theoretical chemistry. Until then, it had been scarcely possible to describe complex structures, such as molecules, using mathematical equations to calculate their behavior. Although the underlying laws were well known, they were too complex to be managed using calculations alone. The calculation of chemical bonds between the atoms of a molecule became possible only after the development of approximation methods and the more ubiquitous use of computers in the 1960s. In chemistry, this development marked the transition from an experimental to a computable science. Two leading fundamental scientists of that time were Walter Kohn and John A. Pople. In 1998, they were awarded the Noble Prize for Chemistry, Kohn for the development of the DFT and Pople for the development of computation methods in quantum chemistry. Since then, computing capacity has multiplied exponentially and DFT has developed further, such that it can now be used for larger and more specific structures.
The Authors:
Christoph R. Jacob has been heading the Junior Research Group for Theoretical Chemistry at the Center for Functional Nanostructures (CFN) of the Karlsruhe Institute of Technology (KIT) since 2010. He studied chemistry and mathematics at the University of Marburg and the University of Karlsruhe (today’s KIT). In 2007, he was conferred his doctorate by the VU University of Amsterdam (The Netherlands). He worked at Auckland University (New Zealand) and ETH Zurich (Switzerland).
Markus Reiher has been a professor of theoretical chemistry at ETH Zurich (Switzerland) since 2006. He studied chemistry, was conferred his doctorate in theoretical chemistry by the University of Bielefeld, and earned his post-doctoral lecture qualification at the University of Erlangen in 2002. He then worked as a private lecturer at the University of Erlangen and the University of Bonn. In 2005, he accepted a professorship for physical chemistry at the University of Jena. A year later, he accepted a call by ETH Zurich.
DFG Center for Functional Nanostructures (CFN)
The DFG Center for Functional Nanostructures (CFN) focuses on an important area of nanotechnology, functional nanostructures. Its excellent interdisciplinary and international research is aimed at representing nanostructures with new technical functions and at making the first step from fundamental research to application. Presently, more than 250 scientists and engineers are cooperating in more than 80 partial projects at the CFN in Karlsruhe. The focus is placed on the areas of nanophotonics, nanoelectronics, molecular nanostructures, nanobiology, and nanoenergy. The building of the CFN is located on the Campus South of the Karlsruhe Institute of Technology.
www.cfn.kit.edu
* First Publication of image 1: Julia Rinck, Ghenadie Novitchi, Willem Van den Heuvel, Liviu Ungur, Yanhua Lan, Wolfgang Wernsdorfer, Christopher E. Anson, Liviu F. Chibotaru, and Annie K. Powell, Angew. Chem. Int. Ed. 2010, 49, Single-Molecule Magnets, p. 7585